Ssu72 Method
Function
The results for the functional component of the analysis were collected from a number of sources. Firstly, a BLAST search was performed using the NCBI database and the 'blastp (protein-protein BLAST)' algorithm (1). Secondly, a literature review was conducted, using the PubMed database (2), on the hits returned from that search. Thirdly, the Drosophila Ssu72 protein, PDB accession number 3FDF_A, was submitted to the ProFunc webserver (3). The automated results returned were then analysed and further searches of PubMed were conducted as necessary - these were dictated by the structural and evolutionary results obtained in their respective sections of this paper.
Structure
-The images were obtained using the RasMol and Dali server. -DaliLite v. 3 was used to find proteins with similar structures and superimpose them, also for Multiple Structure Alignment. -Swiss-model was used for predict the 3-d structure of the Ssu72 RNA polymerase II CTD phosphatase homolog [Homo sapiens]
Evolution
Multiple sequence alignment
NCBI’s psiBLASTp [1] search was run on all of the databases (‘nonredundant’) to yield around 200 significant matches. The search was repeated on the SwissProt database to give 24 matching high quality sequences. The Protein Data Bank yielded no significant matches.
The sequences from the ‘nonredundant’ search were aligned using ClustalX (figure 21). The sequences were well conserved, so a high gap creation penalty (20) was used. Around 30 low matching sequences with large insertions and a few very short sequences were removed, and long sequences were cut to size. (Length was judged relative to the Drosophila sequence of interest.) Closely matched variants from the same organism were removed to reduce redundancy.
The sequences from the SwissProt database were also aligned using ClustalX, with a gap penalty of 20 (figre 20). Eight long sequences were cut to size. Two redundant human sequences were removed. One of the two remaining human sequences placed Homo sapiens with the tetrapods; the other was distant from all the other tetrapods, with a bootstrap of 91% (see figure 25). The first sequence was selected for the tree. It is probable that the second sequence is so divergent because it is inactive. The Cryptococcus Neoformans sequence (Q5KIT2), derived from gene prediction, was too short to show the residues of the active site. It was replaced with a closely matching longer sequence (XM_770627.1) from RefSeq for the alignment. It was not necessary to replace the sequence for the tree, because the missing residues are so highly conserved.
ClustalX was also used to produce an alignment of the target protein and eight phosphatases found by Secondary Structure Matching in Profunc (figures 7 and 22).
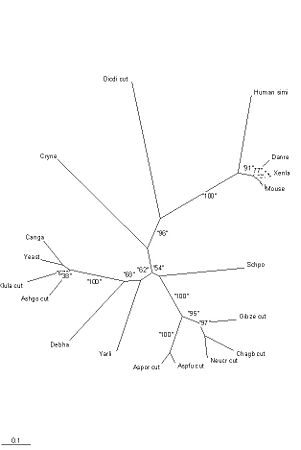
Figure 25: Phylogenetic tree showing the divergence between the second human sequence and the tetrapods.

Phylogenetic tree
The SwissProt sequences were used for the tree (figures 23, 24 and 25). Phylip-3.63 programs were used to generate the tree and bootstrap values (100 replications). The tree’s structure was compared with the taxonomy provided with NCBI’s psiBLASTp results.
Abstract | Introduction | Results | Discussion | Method | References