Ssu72 Results
Function
BLAST results
Running a protein BLAST search on 'serine phosphatase of RNA polymerase II CTD (SSU72 superfamily)' from Drosophila melenogaster (the 'Drosophila Ssu72 protein') returned a large number of highly 'relevant' (bit score > 200) sequences that were of a very similar length (see Figure 1) to that of the query. However, the first 27 of those sequences were not considered relevant for the purposes of this paper for a number of reasons. For instance, some were merely homologous proteins in other closely related Drosophila species that had been identified in a similarly automated manner to this one and for which there were no PubMed publications available and thus no information that could be used to easily infer functionally relevant details. Similarly, other proteins, in more distantly related organisms, had not been functionally characterised by experimentation or structural analysis, presumably because they had been identified by high-throughput sequencing and had seen minimal, if any, human input. Some BLAST hits were labelled as hypothetical proteins and were therefore not necessarily protein coding and biologically expressed. Notwithstanding this, the first relevant protein identified was 'SSU72 RNA polymerase II CTD phosphatase homolog (S. cerevisiae)', the human Ssu72 protein, for which there were a small number of articles available on PubMed (see Figure 2), as well as expression data from SymAtlas (see Figure 3). This protein was also found to have a 60.21% sequence identity with the Drosophila Ssu72 protein, with no gaps in the alignment. Thus, a review of the literature surrounding it was considered a suitable entry-point in the process of this analysis.

Figure 1: BLAST result for the Drosophila Ssu72 protein. Hits highlighted in grey were not considered in this analysis. The human Ssu72 protein is indicated by the black bar.

Figure 2: PubMed results for human Ssu72 protein.
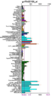
Figure 3: Expression profile for the human Ssu72 protein.
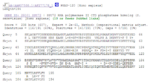
Figure 4: Multiple sequence alignment of Drosophila Ssu72 (query) and Human Ssu72 (sbjct) from BLAST result.




Literature review and ProFunc results
The human Ssu72 protein, for which there exist a number of eukaryotic homologues (Sun and Hampsey 1996), was first proposed to function as a protein tyrosine phosphatase (Meinhart, Silberzahn, & Cramer, 2003). Therefore, given this functional information about an analogous protein, a ProFunc analysis was carried out on our candidate protein, Drosophila Ssu72, with the purpose of comparing components of its primary and secondary structure with proteins that do not necessarily have a high level of overall sequence similarity – thus being unlikely to surface on a non-targeted search of a large database (ie. by BLAST) – but which may have shared catalytic domains. Despite offering a number of powerful results (see Figures 5 - 9), the ProFunc analysis of the Drosophila Ssu72 protein returned just two sets of informative data. The first of these was from the SSM (Secondary Structure Matching) program, which 'identifies proteins in the PDB that have a similar fold to that of the query structure' (Krissinel & Henrick, 2004). The summary of the results of this analysis (see Figures 10 and 11) is by itself promising, as the program exclusively returned proteins which function as phosphatases, all but one of which specifically has phospho-tyrosine as a substrate.
In the next set of results, generated by the so-called 'reverse template' program, the '[target] structure ... is broken up into a large number of templates and these in turn are scanned against a representative set of structures in the PDB' (Laskowski, Watson & Thornton, 2005). In effect, triads of residues from the queried protein are compared against a database of known catalytic sites for matches. The summary of the results (see Figures 12 and 13) show that, while there is only a low level of overall sequence identity (~ 20%) between the target structure and the hits returned by the program, the Drosophila Ssu72 protein shares three important residues - Val12(A), Cys13(A), Ser20(A) - with a number of different protein tyrosine phosphatases and that these residues occupy equivalent coordinates within their respective 3-D structures (see Figures 14 - 16). The implications of these results will follow in the 'function' section of the discussion.

Figure 5: First section of ProFunc results summary.
Figure 6: Second section of ProFunc results summary.

Figure 7: Third section of ProFunc results summary.
Figure 8: Fourth section of ProFunc results summary.

Figure 9: Fifth section of ProFunc results summary.

Figure 10: 10 structural matches returned from the Secondary Structure Matching program.

Figure 11: Alignment of secondary structure schematics from the 10 structural matches returned from the SSM program.

Figure 12: Summary of the reverse template results - certain matches.

Figure 13: Summary of the reverse template results - probable matches

Figure 14: Representative reverse template result - 1

Figure 15: Representative reverse template result - 2

Figure 16: Representative reverse template result - 3










Experimental evidence found in the literature, however, suggests that the human Ssu72 protein functions not as a tyrosine phosphatase, but as a serine phosphatase with specificity to the S5-P of the C-terminal domain of RNA Polymerase II (Krishnamurthy et al., 2004). Further, it also has a role in 3' mRNA processing (He et al., 2003), which is independent of its phosphatase activity (Krishnamurthy et al., 2004). However, there is nothing in the literature which suggests a mechanism by which this specificity to phospho-serine is conferred.
STRING results
Unfortunately, a search of the STRING database did not return any results for the Drosophila Ssu72 protein (see Figure 17). However, a number of interactions were returned when the amino acid sequence of the human protein was searched (see Figure 18). Somewhat disappointingly, most of these results were generated from the 'textmining' component of the search (see Figure 19). However, those papers which were reported under that view of the results will be considered in the discussion of this analysis.

Figure 17: No results for Drosophila Ssu72 protein in the STRING database.

Figure 18: Results for human Ssu72 protein in the STRING database.

Figure 19: Results of the 'textmining' view in the STRING database for human ssu72 protein.


References
He, X.,Khan, A. U., Cheng, H., Pappas Jr D.L., Hampsey M. and Moore, C.L. (2003) Functional interactions between the transcription and mRNA 3′ end processing machineries mediated by Ssu72 and Sub1. Genes Dev. 17, pp. 1030–1042.
Krishnamurthy, S., He, X., Reyes-Reyes, M., Moore, C., and Hampsey, M. (2004) Ssu72 Is an RNA Polymerase II CTD Phosphatase. Molecular Cell 14(3), pp. 387-394.
Krissinel, E., and Henrick, K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions Acta Cryst., D60, 2256-2268.
Laskowsk,i R.A., Watson J.D., and Thornton J.M. (2005). Protein function prediction using local 3D templates. J. Mol. Biol., 351, pp. 614-626.
Sun, Z-W. and Hampsey, M, (1996) Synthetic enhancement of a TFIIB defect by a mutation in SSU72, an essential gene encoding a novel protein that affects transcription start site selection in vivo. Mol. Cell. Biol. 16, pp. 1557–1566.
Evolution
Multiple sequence alignment
The psiBLASTp searches show that sequences homologous to Ssu72 are present in many genomes - many of the results are predicted genes in whole genome annotations. Also, the results indicate that some organisms have more than one copy of the gene- for example, Homo sapiens has a match on chromosome 21 (Gene ID 419410) and on chromosome 11 (Gene ID 390031). Other organisms with multiple copies are Pan Troglodytes (chromosomes 11: 745998 and X: 473738) and Canis familiaris (chromosomes 5: 479567 and 21: 485220).
The SwissProt sequences align well, with occasional insertions/deletions (see figure 20). In particular, the Pezizomycotina share two large additional regions in comparison to Drosophila. Four of the yeasts also share a unique region. (The alignment with the Pezizomycotina region is spurious). Debaromyces hasenii also contains two additional regions.
The regions that form the active site (the catalytic residues and the aspartate loop) are highly conserved. Other conserved regions fall within secondary structure features (helices, sheets, hairpin turns).
An alignment of sequences from all the NCBI databases (figure 21) shows a very similar pattern of conserved residues over 78 different organisms. In particular, the key residues in the active site are either invariant or very highly conserved.
The alignment to the eight phosphatases (figure 22) shows that the catalytic residues are the same in the target protein (3fdf) and the phosphatases. However, the aspartate loop in the tyrosine phosphatases is smaller and further along the sequence. Most of these are sequences of tyrosine (or related) phosphatases, with the exception of the acid phosphatase, 2p4u.

Figure 20: An alignment of the SwissProt sequences, and the target protein, annotated to show the active site regions and the secondary structures corresponding to the conserved regions.
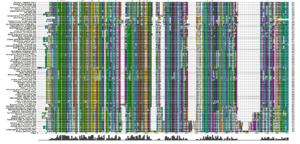
Figure 21: An alignment of sequences from 78 organisms. The active site is conserved, as are many of the regions shown in the annotated SwissProt alignment.

Figure 22: An alignment of eight phosphatases and the target protein. The catalytic site is at 12-20; the aspartate loop is at 142-145 in the target and 173-175 in the phosphatases.



Phylogenetic tree
The tree corresponds perfectly to the current taxonomy (figures 23 and 24). The overall grouping is into three branches: Fungi, Amoebozoa and Metazoa. Clear splits in the taxonomy, such as the one between Danio rerio (zebrafish) and the tetrapods, correspond to very high bootstrap values. Low bootstrap values occur when the organisms cannot be ranked based on this protein sequence alone. In some cases, this is because three groups may really be equidistant: for example, Shizosaccharomyces pombe belongs to neither the Pezizomycotina nor to the Saccharomycetales, and the bootstrap value of 56% indicates that a three-way split between the groups would be more appropriate. In other cases, such as the cluster of four yeasts (Candida glabrate, etc.) and the group of tetrapods (Xenopus laevis etc.), the organisms are too similar to be ranked given this highly conserved protein, and the low bootstrap values reflect this.
The tree contains a variety of organisms. The tree does not have any unusual features that need clarification, but it would still be interesting to include more organisms. In particular, plants are not represented at all. However it seems likely from these results that even with additional sequences the tree will still follow known phylogeny.
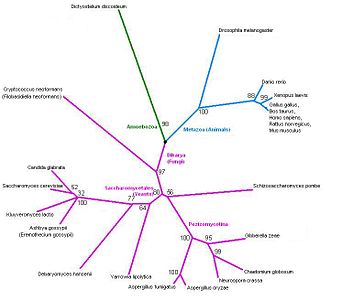
Figure 23: Phylogenetic tree.
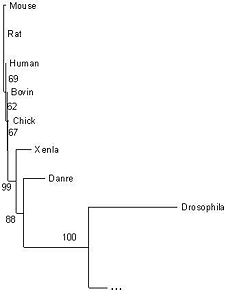
Figure 24: A closer view of the Metazoa.


Structure
Structure determined using XRD. The following images were obtained using RasMol.
PDB 3fdf protein - Secondary Structures

PDB 3fdf protein - Chains

PDB 3fdf protein - Molecular surface

Protein Structure Database Searching by DaliLite v. 3
The Dali server is a network service for comparing protein structures in 3D. You submit the coordinates of a query protein structure and Dali compares them against those in the Protein Data Bank (PDB).
These are the chains of different molecules that best matched to the Ssu72 RNA polymerase II CTD phosphatase homolog (it is important to notice that the first match corresponds to the protein that is being researched).
PDB ID ... Z rmsd lali nres %id PDB Description
3fdf-A..... 37.3 0.0 191 191 100 PDB MOLECULE: FR253;
3fmv-H... 32.4 0.6 190 190 100 PDB MOLECULE: SERINE PHOSPHATASE OF RNA POLYMERASE II CTD;
1dg9-A... 11.4 2.3 120 157 13 PDB MOLECULE: TYROSINE PHOSPHATASE;
1rxi-A...... 9.9 2.4 109 131 15 PDB MOLECULE: ARSENATE REDUCTASE;
DaliLite results: Multiple Structure Alignment


DalaLite results: Superimposed structures
FR253-TYROSINE PHOSPHATASE

FR253-ARSENATE REDUCTASE

FR253-SERINE PHOSPHATASE OF RNA POLYMERASE II CTD

Secondary Structures
FR253

SERINE PHOSPHATASE OF RNA POLYMERASE II CTD

TYROSINE PHOSPHATASE

ARSENATE REDUCTASE

3fdf vs 1dg9
Drosophila

Tyrosine Phosphatase

STRING - Known and Predicted Protein-Protein Interactions


Homo sapiens homolog
Structure obtained through Swiss-Model Automatic Modelling Mode

Superimposition, using Dali server, between Homo sapiens homolog and 3fdf-A

Structure Alignment


Heading
Abstract | Introduction | Results | Discussion | Method | References