COASY results: Difference between revisions
| Line 161: | Line 161: | ||
Image:COASYHydro2.jpg|'''Figure 6 '''<BR> Coenzyme A Synthase Mus. musculus ribbon structure showing hydrophobic regions. | Image:COASYHydro2.jpg|'''Figure 6 '''<BR> Coenzyme A Synthase Mus. musculus ribbon structure showing hydrophobic regions. | ||
</gallery> | </gallery> | ||
===Functional Sites Found By Pattern Search=== | |||
===Functional Sites Found by Sequence Conservation In Structurally Related Proteins=== | |||
===Functional Sites Found by Structure Conservation In Structurally Related Proteins=== | |||
===Functional Sites Found Through ACO crystallization=== | |||
Revision as of 18:28, 9 June 2007
Structure of Coenzyme A Synthase
Conezyme A Synthase is structurally composed of seven strands, eleven helices and thirteen beta turns (EMBL EBI, 2005) (see Figure 2). Analysis of structurally related proteins (Holm & Sander, 1993) showed a trend for transferase (RCSB, 2007) class proteins with Rossmann class folds (Rossmann, 1973). The Rossmann topologies of fold’s are an alpha-beta class fold that forms a three or more layer beta strand sandwich alternating with alpha helices (beta-alpha-beta-alpha-beta). PFAM classification placed these into either the Cytidylyltransferase or Dephospho-CoA kinase familes, matching the two domains of Coenzyme A Synthase. Those classified under the Dephospho-CoA kinase type were commonly of the P-loop containing nucleotide triphosphate hydrolases (Sanger Institute, 2005) homology whilst those classified as Cytidylyltransferase were of the Tyrosol-Transfer RNA Synthetase (Sanger Institute, 2005). As the majority of the structurally related proteins identified contained the DPCK domain solely, and the motif for a P-loop (Table 1) was identified in the Conzyme A Synthase sequence (Bairoch, Bucher, & Hofmann, 1997) it is suggested that Coenzyme A Synthase is also of the P-loop containing nucleotide triphosphate hydrolases homology of folds.

The secondary structure of Mus musculus with indicated ligand interaction sites (EMBL EBI, 2005).
Table 1
| Pattern-ID: | GLYCOSAMINOGLYCAN PS00002 PDOC00002 |
| Pattern-DE: | Glycosaminoglycan attachment site |
| Pattern: | SG.G |
| Position: | 84 SGSG |
| Pattern-ID: | PKC_PHOSPHO_SITE PS00005 PDOC00005 |
| Pattern-DE: | Protein kinase C phosphorylation site |
| Pattern: | [ST].[RK] |
| Position: | 3 SDK |
| Position: | 52 SFR |
| Position: | 86 SGK |
| Pattern-ID: | CK2_PHOSPHO_SITE PS00006 PDOC00006 |
| Pattern-DE: | Casein kinase II phosphorylation site |
| Pattern: | [ST].{2}[DE] |
| Position: | 39 SHNE |
| Position: | 251 TLWE |
| Position: | 260 SQVE |
| Pattern-ID: | MYRISTYL PS00008 PDOC00008 |
| Pattern-DE: | N-myristoylation site |
| Pattern: | G[^EDRKHPFYW].{2}[STAGCN][^P] |
| Position: | 82 GISGSG |
| Position: | 222 GLSEAA |
| Pattern-ID: | ATP_GTP_A PS00017 PDOC00017 |
| Pattern-DE: | ATP/GTP-binding site motif A (P-loop) |
| Pattern: | [AG].{4}GK[ST] |
| Position: | 82 GISGSGKS |
| Pattern-ID: | UPF0038 PS01294 PDOC00996 |
| Pattern-DE: | Uncharacterized protein family UPF0038 signature |
| Pattern: | G.[LI].R.{2}L.{4}F.{8}[LIV].{5}P.[LIV] |
| Position: | 136 GTINRKVLGSRVFGNKKQMKILTDIVWPVI |
Localisation Expression of Coenzyme A Synthase
Baseline expression of CoAsy was present in all tissues. Expression was slightly elevated in adipose tissue, and highly elevated in the liver and kidney (See Figure 3). Baseline expression of the other enzymes involved in the CoAsy pathway was also present in all tissues. However, expression of these enzymes was otherwise unlike that of CoA, as they were not particularly elevated in the liver and kidney (data not shown).

CoA Synthase Expression. Reproduced from Genomics Institute of Novartis Research Foundation (2007).
Domain and Structural Analysis
Local sequence alignment showed that CoAsy Chain A residues 13-281 corresponded to residues 295-563 on full length CoAsy. This indicates that a 295 amino acid stretch extending from residues 1 to 295 on the full length protein was cut out during structural analysis and was not present on chain A.
Bioinformatic analysis using NCBI Entrez protein indicated that mouse CoAsy (full length) possess two domains, a PPAT and a DPCK domain. The PPAT domain extended from residues 194-338, and the DPCK domain from residues 359-536. PFAM analysis of CoAsy chain A also indicated the presence of two domains, the DPCK domain from residues 64-242 and a CTP transferase 2 domain (part of the cytidylyltransferase family, of which PPAT is a member) extending from residues 1-44, which corresponds to a fragment of the PPAT domain. The position of these domains on CoAsy chain A is shown in Figure 4. Profunc analysis of CoAsy chain A detected the DPCK domain, but not the fragment of the PPAT domain.

Surface structure of Mus musculus Coenzyme A Synthase with Phosphopantetheine adenylyltransferase (PPAT) and Dephospho-CoA kinase (DPCK) domains marked in red and blue respectfully.
Structural Elements and Functional Binding Sites of Coenzyme A Synthase
Comparison to CoAsy’s closest structurally related protein, Dephospho-CoenzymeA Kinase, finds that whilst it shares only twenty percent of it’s sequence, it is structurally very similar (Figure 3). Apart from the lack of a PPAT domain on the Dephospho-CoenzymeA Kinase molecule (see Figure 7 for comparative location of PPAT), all structural features are roughly the same between the two proteins. As helices, sheets and loops stay relatively constant between the two, and Dephospho-CoenzymeA Kinase has a related function to CoAsy it can be assumed that the location of the ATP binding site on the DPCK doimain of CoAsy is the same as that on Dephospho-CoenzymeA Kinase.

Electrostatic Surface Potential
Analysis of electrostatic surface potential maps for CoAsy finds that there is a strongly positive charged region, between residues 70 and 80, on the first helix that lines up with the proposed ATP binding site (Figure 4). It can also be noted that the inner wall of the major cleft alternates strongly between a positive and negative charge and this lines up with the ACO ligand that is included in the 2F6R PDB entry at this location (EMBL EBI, 2005).

Surface map of Mus musculus Coenzyme A Synthase with electrostatic charge indicated, ATP ligand is included at P-Loop bind site of DPCK domain and ACO at the proposed Dephospho Coenzyme A Kinase cleft.
Residue Flexibility
Structural analysis of Coenzyme A Synthase B-Factor scores finds it to be majoritly stable close to the center, and on the N-terminal of the protein. The most flexible regions are on the eighth helix and on the outer edges of the protein (Figure 4). The position of most flexibility on the protein is at the top of the eight loop, on the DPCK domain, and this lies between both the bind site for ATP and the location of ACO on the 2F6R PDB entry (EMBL EBI, 2005).

Coenzyme A Synthase Mus musculus surface structure marked by B-Factor.

Ribbon representation of Mus musculus Coenzyme A Synthase (RCSB, 2007) with conservation of structural features marked for structurally related proteins. Note P-Loop conservation (residues 65-85).
Hydrophobicity
Hydrophobic mapping of Mus. Musculus Coenzmye A Synthase finds a number of hydrophobic stretches including the inner strands and the N terminal Figure 5. A study by (Zhyvoloup, et al., 2003) also found that the Mus. Musculus CoAsy had a hydrophobic N terminal and that this is a conserved feature for CoAsy across a broad number of species.
- Figures 6 & 6

Figure 6
Sequence alignment of N domain (first 30 residues) of Coenzyme A Synthase proteins (bottom) with hydrophobic residues shaded. The hydrophobicity profile (top) of Coenzyme A Synthase was generated by TMpred program (Hofmann & Stoffel, 1993). A Modular representation of Coenzyme A Synthase is presented in the middle (Zhyvoloup, et al., 2003).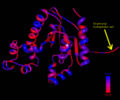
Figure 6
Coenzyme A Synthase Mus. musculus ribbon structure showing hydrophobic regions.


Functional Sites Found By Pattern Search
Functional Sites Found by Sequence Conservation In Structurally Related Proteins
Functional Sites Found by Structure Conservation In Structurally Related Proteins
Functional Sites Found Through ACO crystallization
Abstract | Introduction | Results | Discussion | Conclusion | Method | References